GPIアンカーとは?
細胞表面にはGPI(glycosyl-phosphatidyl-inositol)と呼ばれる糖脂質によって細胞膜に結合するタンパク質のグループ(GPIアンカー型タンパク質)が発現している。GPIの基本構造はホスファチジルイノシトール(PI)、グルコサミン(GlcN)、3つのマンノース(Man)、2つのエタノールアミンリン酸 (EtNP)から成り立っており(図1)、複数のステップにより合成される。ほ乳類においては現在までに150種以上のGPIアンカー型タンパク質(GPI-APs)が知られており、酵素や受容体、接着因子、補体制御因子など個体発生や神経発達、免疫機能、受精等非常に重要な働きを担っている。GPIアンカーが正しい構造で、かつ細胞に必要な量が生合成されないと、GPI欠損症と呼ばれる病気を発症する。我々は、GPIアンカー型タンパク質の生合成経路や機能を解析し、その不調によって起こるGPI欠損症の病態を解明し、診断、治療に繋げるべく研究を展開している。

図1. GPIアンカー型タンパク質の構造
GPIアンカー型タンパク質の生合成と輸送
1) GPIアンカーの生合成
GPI-APは小胞体(ER)で蛋白部分とGPI部分が別々に合成される。GPI生合成の最初のステップは触媒サブユニットPIGAとPIGC, PIGH, PIGP, PIGQ, PIGY, DPM2の7個のタンパク質からなる酵素複合体によって担われている。その後10個のステップを経て完成したGPI-APに付加される。この反応は触媒サブユニットであるPIGKとGPAA1、さらにPIGS, PIGT, PIGUの5個のタンパク質からなるトランスアミダーゼ複合体によって担われ、この酵素はGPI付加シグナルを持つタンパク質のC末端シグナルペプチドを認識して切断しGPIの末端のエタノールアミンのアミノ基と共有結合させる。これらのように小胞体でGPI-APに付加されるまでのステップに関わる遺伝子群をPIG (Phosphatidyl Inositol Glycan) genesと呼びほぼクローニングされた順にアルファベットの名前がついている。またその後の修飾に関係する遺伝子群をPost GPI Attachment to Proteins (PGAP) genesと呼んでいる (J Biochem 2008)。最近になって、2つめのGPI中間体であるグルコサミンPIを小胞体の細胞質側から内腔に移すスクランブラーゼ, CLPTM1Lを同定した(PNAS 2022)。
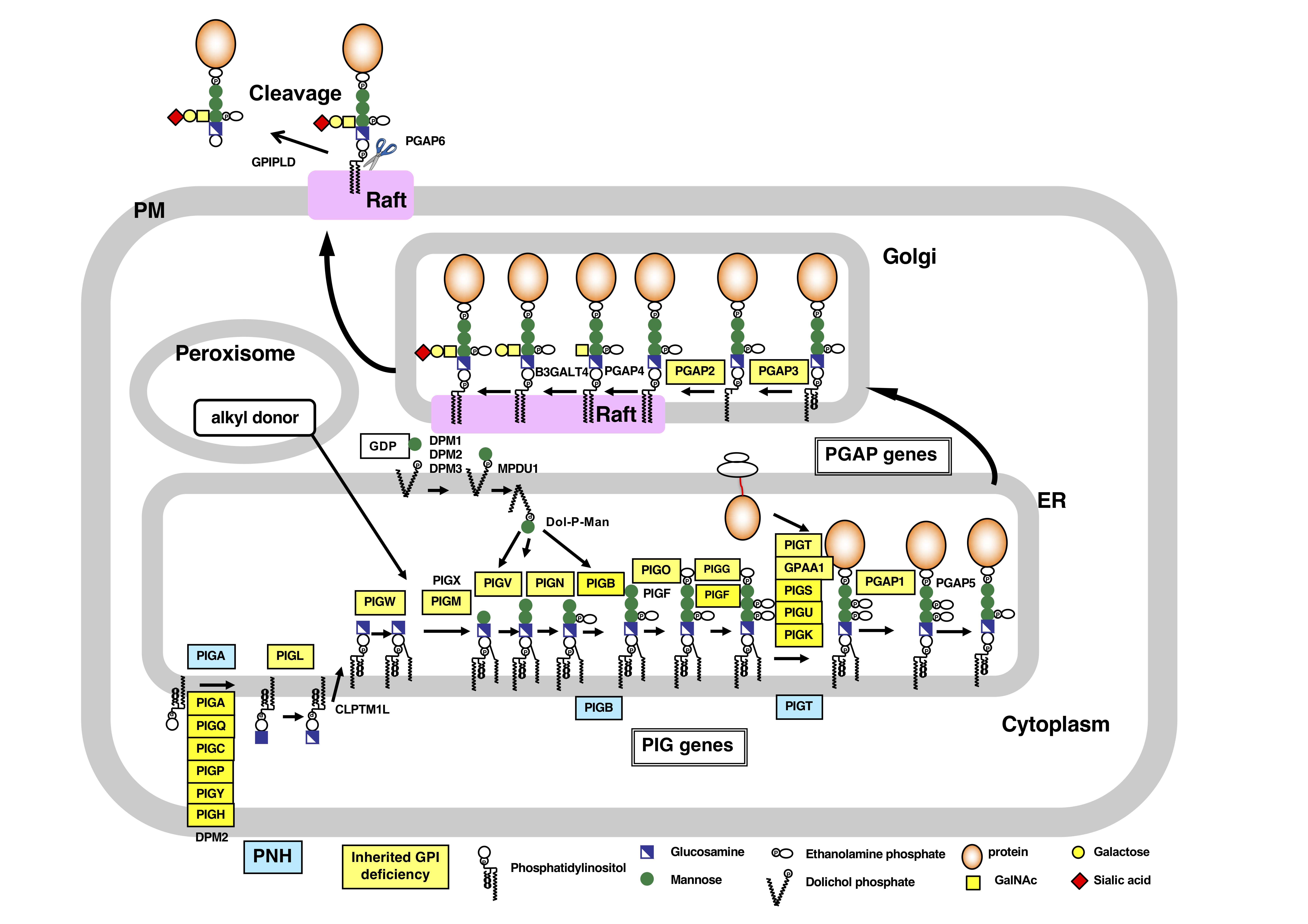
図2. GPIアンカー型タンパク質の生合成と輸送
2) GPIアンカー型タンパク質のリモデリング
・イノシトールの脱アシル化
GPI生合成の初期にPIGWによってGlcN-PIのイノシトールにアシル基が付加される。この反応はGPIの生合成が細胞質側から小胞体内膜側に移行した直後に起こり、その後の効率的なGPI生合成に必要である。このアシル基はGPIアンカーがタンパク質に付加された後に小胞体でリパーゼモチーフ(GxSxG)を有するタンパク質、PGAP1によって除かれる。この遺伝子の欠損細胞では細胞表面にはアシル基の付いた異常構造のまま発現する (J Biol Chem 2004)。
・GPI糖鎖リモデリング(側鎖EtNPの除去)
GPI生合成過程において3つのManにはそれぞれEtNPが付加される。このうちPIGG/PIGF複合体によって2つ目のManに付加されたEtNPはGPIアンカーがタンパク質に付加された後に小胞体でPGAP5によって除かれる。このステップは小胞体からゴルジ体への効率的な輸送に必要である(Cell 2009) 。
・脂肪酸リモデリング
この反応はゴルジ体で行われ、GPI-APsが脂質ラフトに局在するために必須である。まず脂質部分のsn-2位に付加されている不飽和脂肪酸(アラキドン酸(C20:4)が主要)がPGAP3により除去され、飽和脂肪酸(ステアリン酸(C18:0))が付加される。この反応にはPGAP2が必須である。PGAP3の欠損細胞ではGPI-APsはリモデリングを受けない異常構造のまま細胞表面に発現するが、脂質ラフトに局在できない。PGAP2の欠損細胞ではGPI-APsはsn-2位に脂肪酸が付加されないリゾ体のまま細胞表面に運ばれるがリパーゼによって切断遊離され、その結果細胞表面のGPI-APsは著減する(Mol Biol Cell. 2006, 2007)。
・細胞膜からの遊離
GPI-APであるCripto-1は発生初期に発現し形態形成に重要なタンパク質であるが、Phospholipase A2であるPGAP6によりリゾ体になり細胞膜から遊離しNodal のリガンドとしてトランスに機能する。PGAP6ノックアウトマウスは胎生早期で致死となりCripto-1のノックアウトマウスと同じ表現型を示したことから、この細胞膜からの遊離が発生初期に重要であることがわかった(J Cell Biol. 2016)。
3) GPIアンカー型タンパク質の側鎖
多くのGPI-APはゴルジ体においてPGAP4によって1つ目のManの4位にNアセチルガラクトサミン(GalNAc)が付加される(Nat. Commun. 2018)。この側鎖にガラクトースがB3GALT4(Beta-1,3-Galactosyltransferase 4)によって付加され(Nat. Commun. 2020)、さらにシアル酸が結合した構造が知られている。この構造は神経系の組織に多く見られることから神経機能に重要であると考えられる。また一部のGPI-APにはPIGZによって3つ目のManの側鎖としてさらに4つ目のManが付加されることが知られている。
今後の取り組み
GPI-APの生合成と輸送に関与する経路は上記のようにほぼ全容が解明され、あと1〜2カ所を残すのみとなっている。しかしながらどのように生合成や輸送が調節されているか、あるいはその品質管理や分解経路についてはまだまだ不明な点が多い。生合成の最初のステップと最終のステップはいずれも複数の因子の酵素複合体が機能しているが個々の因子の役割は不明である。これらの解明が生合成や輸送の調節機構や分解経路を解析する糸口になると考えている。また神経系の組織に多く見られるGPI-APのGalNAcの側鎖の機能的意義についても解析を進めている。
GPIアンカー欠損症
体細胞突然変異による後天的な疾患と生殖系列の変異によって起こる先天性の疾患がある。
1) 発作性夜間ヘモグロビン尿症(paroxysmal nocturnal hemoglobinuria: PNH)
溶血性貧血、骨髄不全、深部静脈血栓を3主徴とする血液疾患である。溶血発作のエピソードと末梢血のフローサイトメトリーで顆粒球および赤血球表面のGPI-APsであるCD59やDAFの発現が低下あるいは欠損している細胞集団を確認することで診断される。後天的に1個あるいは数個の造血幹細胞のPIGA遺伝子に突然変異が起こってGPI欠損細胞となり、クローナルに増殖することによって発症する疾患なので正常細胞とGPI欠損細胞が混在していることが特徴である。
PNH のGPI欠損赤血球は補体制御因子であるCD59やDAFを欠損しているため、細胞表面での補体の活性化を制御することができないので感染等に伴って血管内で補体の活性化が起こったとき,異常細胞は一斉に溶血し,溶血発作となる。抗C5モノクローナル抗体医薬のエクリズマブの投与により補体の活性化が抑制され患者のQOLは劇的に改善している。
今までPIGAの変異のみがPNHの原因遺伝子とされてきた。その理由はPIGAが唯一のX染色体遺伝子で女性も男性も機能的なアレルが1つで1ヒットの体細胞突然変異で細胞はGPI 生合成能を失うからである。他の遺伝子は常染色体遺伝子で二つのアレルが機能しているので二つの体細胞変異が重ならないとGPI 欠損細胞にならず、その確率はきわめて低い。しかしながら最近、次世代シークエンサーを用いた解析によってPIGTを責任遺伝子とする国内症例を含む4例のPNHが見つかった(Blood 2013, J. Clin. Invest 2019)。4症例すべてで遺伝的に1本のアレルに変異があるところに、造血幹細胞において体細胞突然変異が起こりPIGT周辺領域の欠損が起こって発症したものであった。これらはPIGA-PNHとは異なり、溶血発作に加えて髄膜炎、関節痛、発熱などの自己炎症症状が特徴的であった。PIGT欠損細胞ではフリーGPIが細胞膜上に発現しており、それが補体活性化と重なってインフラマソームの活性化を来すことがわかった(J. Clin. Invest 2019)。さらにオランダとの共同研究によりPIGBを責任遺伝子とするPNHが見つかり(Blood Adv. 2020) 、やはり自己炎症症状を来していた。母親由来の生殖系列の変異が造血幹細胞の突然変異でCN-LOH( copy number neutral-loss of heterogeneity)が起こり、発症していたが母親にもPNHクローンが見られ初めての家族例である。 このようにまれではあるが他の遺伝子を責任遺伝子とするPNHが今後見つかる可能性がある。
2) 先天性GPI欠損症(Inherited GPI deficiency: IGD)
2006年に最初のIGD症例であるPIGM 欠損症を報告したが(Nat Med. 2006)、2010年から次世代シークエンサーによる解析で次々見つかっており、国内では約50例、海外合わせて400例以上の報告例があるが、今後も疾患の周知に伴って診断例が増加すると思われる。2016年より厚労省の研究班を立ち上げ、2017年度より指定難病、2018年度より小児慢性特定疾病に認定されている。
前述のようにGPI-APの生合成と修飾には29個の遺伝子が関わっている。IGDはこれらの遺伝子の変異により起こる遺伝性の疾患で、現在24個の遺伝子変異による症例が報告されている(図2)(脳と発達2015)。GPIアンカーが欠損すると150種類以上の重要なタンパク質が細胞表面に発現できず細胞内で破壊されたり、異常な構造で発現したりして生体は大きな影響を受ける。GPI生合成の最初のステップに必要な遺伝子PigaのノックアウトマウスはGPIアンカーが完全に欠損するので胎生致死になる。従ってIGD症例の多くは遺伝子変異によって活性の低下した部分欠損症である。
IGDではどのステップの遺伝子の変異か、またその変異によってタンパク質の機能がどの程度低下するかで症状やその重症度が変わる。最も軽症例でも知的障害は必発で、重症度が増すに従って、運動発達障害、てんかん、テント状の口などの異常顔貌、手指・足趾の末節骨や爪の欠損、難聴、ヒルシュスプルング病、心奇形、横隔膜ヘルニア等多臓器の奇形、魚鱗癬などの皮膚症状、目の異常、脂質代謝異常等が見られる。乳児早期発症のてんかん性脳症である大田原症候群やウエスト症候群を呈する場合もある。また重症例では脳MRIで脳幹部や中脳に拡散強調やT2強調画像で高信号を来すことが特徴で、これは病態に代謝異常が関与していることを示唆している。GPI-APであるアルカリホスファターゼ(ALP)の発現低下によりビタミンB6の脱リン酸化が減少した結果細胞内への取り込みが低下し、細胞内ビタミンB6欠乏が起こりこれを補酵素とするアミノ酸代謝の障害が起こっていると考えられる。症例によってはてんかん発作がビタミンB6欠乏によるGABA合成の低下に起因しており、リン酸化のないビタミンB6の大量投与が効果的である(Neurology. 2013)。また小脳萎縮や大脳白質変性が生後も進行する。
次世代シークエンサーの普及により多くの家族性の疾患の原因遺伝子が同定されるようになってきたが、いくつかの疾患がIGDの1病型であるとわかってきた。Hyperphosphatasia Mental Retardation Syndrome (Mabry Syndrome)は古くから高ALP血症と知能障害をきたす劣性遺伝の疾患として知られていたが、遺伝子解析によりPIGV, PIGO, PIGW, PIGL, PGAP2, PGAP3欠損症、つまりIGDのうち高ALP血症を来す病型に一致することがわかった。またFryns syndromeは横隔膜ヘルニアに手指.足趾の末節骨や爪の欠損を伴う疾患であるが、PIGN, PIGV, PIGO欠損症であることが報告されIGDの最重症型と重なることがわかった。CHIME症候群(コロボーマ-先天性心奇形-魚鱗癬型皮膚症-精神遅滞-耳介奇形症候群)もPIGL欠損症であることがわかり、これもIGDの1型であると考えられる。
多くのIGD症例は血液のフローサイトメトリー解析(FACS解析)での顆粒球のCD16発現低下でスクリーニングすることができ、さらに遺伝子解析で責任遺伝子を同定することにより確定する。現在は早期診断法の確立と治療法の開発に力を注いでいる。前述の基礎研究の成果が糸口になる可能性もあり、また症例から新たに発見される現象もある。基礎研究と臨床研究を並行して進めることにより最終的には治療法の開発に繋がる研究をしたいと考えている。
